Primary immunodeficiency
diseases (PIDs) are intrinsic defects of the immune system. Patients
with PID have increased susceptibility to recurrent and persistent
infections, but other symptoms are also common.
Adaptive immune mechanisms recognize and neutralize foreign
molecules or microorganisms in a specific manner. Lymphocytes, B and
T cells, can respond selectively to thousands of non-self materials.
Adaptation is further acquired with memory of previous infections.
Immunodeficiencies impair the functioning of the immune system. Deficiencies
are highly variable with regard to symptoms, phenotype, genotype, severity,
etc, because many cells and molecules are required for both natural
and adaptive immunity.
However, increased susceptibility to infections is common to all immunodeficiencies.
I Classification
More than 70 primary immune
deficiencies (PIDs) are known, and can be grouped according to
the components of the immune
system affected.
1) Antibody deficiency disorders are defects in immunoglobulin-producing B
cells.
2) T cell deficiencies affect the function in killing infected cells
or helping other immune cells.
3) T cell deficiencies result usually in combined immunodeficiencies
(CIDs), where both T
cells and antibody production are defective.
Other IDs affect the
4) complement system
or
5) phagocytic cells
impairing anti-microbial immunity.
Secondary immunodeficiencies may cause similar infections to PIDs,
but secondarily to some other pathological condition such as malnutrition,
age, drugs, tumours, or infections including HIV in
AIDS.
II Incidence
Most PIDs are relatively rare disorders. The incidence of PIDs varies
greatly from about 1:500 births with selective IgA deficiency
to only a few known cases for the rarest disorders.
Table of prevalences for
immunodeficiencies.
III Symptoms
Patients with antibody deficiencies are especially susceptible to encapsulated
bacteria, which cause pyogenic infections. T cell immunodeficiencies
and severe combined immunodeficiencies (SCIDs) are marked with opportunistic
infections caused by common environmental microorganisms. PID patients
have recurrent, serious infections starting early after birth. Children
are protected for 6-12 months by the maternal IgG. Life threatening
symptoms can arise already within the first few days of life in SCID.
IV Treatment
Infections are treated with antibiotics. PID patients require prolonged
treatment with high doses. Antibody deficiencies are treated with intravenous
immunoglobulin substitution therapy. Gammaglobulins are extracted from
human blood from donor pools. Leukocytes are produced in stem cells
in bone marrow. In SCIDs bone marrow transplantation is
the most effective treatment. In certain metabolic disorders ( ADA and PNP deficiency)
enzyme substitution therapy can be applied.
1. Genetics
The immune
system consists of a large number of molecules and processes, and
immunodeficiencies can therefore be caused by genetic alterations at
many loci. A particular PID can be caused by defects in any one of
several molecules that are required for certain responses, because
a defect in any of the sequential steps can impair the system. The
inheritance of the majority of the PIDs is autosomal recessive (AR),
although the most studied cases are X-linked.
Recognition of the enormous range of non-self substances is the basis
of adaptive immunity and
is achieved by mechanisms that produce largely heterogeneous receptors,
namely antibodies, T cell receptors (TCRs), and the components of the
major histocompatibility complexes (MHCs). These molecules owe their
high variability to
a large number of genetic segments, which can be joined in random fashion.
2. B cell immunodeficiencies
B cell immunodeficiencies are antibody deficiency disorders that are
restricted to antibody function. Either B lymphocyte development is
impaired, or B cells fail to respond to T cell signals. All or only selected subsets
of immunoglobulins may be deficient. Patients have recurrent pyogenic
infections with encapsulated bacteria. Infections require early and
vigorous treatment with antibiotics and life-long immunoglobulin replacement
therapy.
2.1 X-linked agammaglobulinemia (XLA)
          
XLA is a typical antibody deficiency in which production of antibodies
is prevented due to a block in the B cell maturation. The prevalence
is about 1:200,000. Serum concentrations of IgG, IgA, and IgM are markedly
reduced. Levels of circulating B lymphocytes are significantly decreased
and plasma cell are absent from lymph nodes and bone marrow, but the
number of T cells is normal or even increased. The clinical phenotype
may be variable and even members of the same family can have different
symptoms. Patients with XLA have
normal response to viral infections and normal V(D)J rearrangement. XLA is
caused by a block in the B cell differentiation. Btk, the affected
protein, is a tyrosine kinase that regulates activity of signalling
pathways by phosphorylation.
Typically, about one third of X-linked PIDs are new, sporadic cases,
where the mutation has appeared in the patient. Bruton tyrosine kinase
(Btk) is the defective molecule in X-linked agammaglobulinaemia (XLA).
Mutations in the gene for BTK prevent B cell maturation since
Btk is crucial signalling molecule regulating B cell development into
antibody-producing plasma cells. Btk protein consists of five domains,
all of which can be affected by disease-causing alterations. The majority
of the known mutations (altogether more than 300) lead to truncation
of the protein due to either nonsense or frameshift mutations because
of insertions, deletions or splice site defects. Although the mutations
are evenly distributed in the BTK gene, the most mutable single
sites in many genetic disorders, including XLA,
are CpG dinucleotides.
In addition to the catalytic tyrosine
kinase domain, Btk contains modules for protein-protein interactions,
namely Tec homology (TH), Src homology 2 (SH2), and SH3 domains. Pleckstrin
homology (PH) domain is responsible for membrane localisation of the
protein by binding to the phosphatidyl inositol groups of membrane
lipids. Btk has several signalling partners. The specificity of the
kinase domain may be increased by simultaneous binding of the SH2 and/or
SH3 domain into their recognition sites in the substrate. The TH domain
binds Zn2+, which is essential for stability. Btk is activated by
stepwise phosphorylation, first by Src family kinases and then by autophosphorylation.
Although many of the mutations cause truncation of the Btk protein,
there are also other types of alterations. For example, a number of
structural mutations lead into misfolded proteins. Three-dimensional
structures of the Btk domains have been used to describe the consequences
of the disease-causing mutations. Of the amino acid-changing missense
mutations, many alter structurally of functionally significant conserved
residues.
2.2 Hyper-IgM syndrome (HIM)

HIM syndrome represents a group of related diseases, the majority of
which are X-linked. XHIM is caused by a genetic defect in the gene
for the CD40 ligand. The patients have severely reduced IgG, IgA, and
IgE serum levels, but normal or even raised IgM levels. In XHIM there
is a failure in heavy chain class switch from IgM to IgG and IgA. Interaction
between CD40L in T cells and CD40 on B cells is a key signal for the
generation of memory B cells and for the formation of germinal centres.
Defective CD40L prevents production of certain antibodies. CD40 is
also involved in interaction with macrophages and dendritic cells,
which induces IL-12 secretion and thereby elicits an immune response
to intracellular microorganisms. Infections in patients with HIM are
similar to those in XLA,
except for higher tendency for persistent or recurrent neutropenia
and thrombocytopenia.
CD40 belongs to the tumour necroris factor (TNF) superfamily of transmembrane
proteins. Signalling through CD40 activates phospholipase Cγ 2
and phosphatidylinositol-3 kinase, upregulates CD23, CD54, and CD80,
and induces transcription factors such as nuclear factor-κ
B. The XHIM causing mutations can appear in any of the domains of CD40L
(intracytoplasmic, transmembrane, extracellular unique, and TNF homology
regions). However, mutations cluster into the TNHF domain. The most
common alterations are point mutations, which typically result in truncation
of the polypeptide chain.
2.3 IgA deficiency
IgA deficiency can affect only IgA levels, or it may be combined with
the lack of other isotypes. IgA deficiency is the most prevalent PID
(1:500 Caucasians), but its mechanism remains unknown. Only about one
third of the patients are particularly prone to infections. The serum concentrations
of the other immunoglobulins are usually normal, but patients have
a high incidence of autoantibodies.
2.4 Selective IgG subclass deficiencies
Selective deficiencies of IgG subclasses,
with or without IgA deficiency, are caused by defects in several genes.
IgG2 deficiency is most common in children, whereas adults more often
have low levels of IgG3.
2.5 Common variable immunodeficiency (CVID)
Common variable immunodeficiency (CVID) is a group of undifferentiated
disorders with defective antibody formation. The incidence is approximately
1:25,000. Patients with CVID usually have normal number of circulating
B cells, but low serum levels
of IgG and IgA. However, the B cells are defective. CVID affects both
females and males equally and it usually has later age of onset than
other antigen IDs. Patients have an unusually high incidence of lymphoreticular
and gastrointestinal malignancies and the incidence of autoimmune disorders
is also increased. CVID forms arise from several different genetic defects.
2.6 Other B cell deficiencies
Other B cell deficiencies have been described including, for
example μ heavy chain deficiency; λ
5 surrogate
light chain deficiency; and κ light chain deficiency.
3 T cell immunodeficiencies
T cell deficiencies usually affect also other components
of the immune system and lead into CIDs.. Patients have a progressive
decrease in the number of circulating T cells, whereas numbers of circulating
B cells and serum immunoglobulin are usually normal. Autoimmunity is
a frequent complication in T cell IDs. In T cell deficiencies bone
marrow transplantation is
in many cases the only long-lasting therapeutic option.
3.1 Purine nucleoside phosphorylase (PNP) deficiency
Purine nucleoside phosphorylase (PNP) deficiency is characterized by
accumulation of toxic purine metabolites, primarily dGTP, in cells.
PNP catalyses the phosphorolysis of the purine nucleosides, (deoxy)inosine
and (deoxy)guanosine, to purine bases and ribose-1-phosphate. dGTP
is particularly toxic to T
cells by inhibition of ribonucleotide reductase and further DNA
synthesis and proliferation. PNP deficiency is often accompanied by
neurologic disorder. The enzyme following PNP in the purine catabolism
is adenosine deaminase, mutations of which cause SCID.
3.2 Zap-70 deficiency

T cell activation triggers cascades of reactions. Zap-70 (ζ -associated
polypeptide of 70 kDa) is a tyrosine kinase that binds with its SH2
domains to the TCR's phosphorylated immunoreceptor tyrosine-based activation
motif (ITAM) sequences. In Zap-70 deficiency,
signalling through TCR is defective, influencing T cell development.
3.3 X-linked lymphoproliferative (XLP) disease
In X-linked lymphoproliferative disease (XLP) or Duncan's disease,
patients are exceptionally susceptible for Eppstein-Barr virus (EBV).
EBV infection causes mononucleosis by vigorous uncontrolled expansion
of both T and B cells. The disease is associated usually either with
hypogammaglobluninaemia, Burkitt lymphoma, carcinoma, some forms of
Hodgkin disease, or several of them. The mortality is complete by the
age of 40 years. SAP (SLAM-associated protein) (also called for DSHP
and SHD1A) is a short SH2 domain containing molecule. SLAM (signalling
lymphocyte activation molecule), also known as CDw150, appears on the
surface of T
cells, where is has a crucial function in stimulation. Phosphorylation
of SLAM provides docking sites for SH2 domain containing proteins including
protein phosphatase SHP-2. SAP regulates the binding by competing for
the SLAM. Mutations in SAP affect the interaction between T and B cells
and leads into uncontrolled B cell proliferation in EBV infection.
SAP mutations either truncate the protein or affect the folding or
recognition of ligand(s) by its SH2 domain.
3.4 Nezelof syndrome
Nezelof syndrome is a T cell deficiency with little or no abnormality
of gammaglobulins. The patients have very small thymuses. Also antibody
synthesis is impaired and IgA can be deficient, whereas IgD or IgE levels
can be elevated. Nezelof syndrome is the most likely PID to be confused
to AIDS.
3.5 CD3ε
and CD3γ deficiencies
CD3 is a multicomponent T cell complex formed of non-identical subunits
that interact with TCR.
Interaction with antigen activates cytokine release and cell proliferation.
Rare CD deficiencies are caused by mutations in the γ and ε subunits.
4 Severe combined immunodeficiencies (SCIDs)
In combined B and T cell immunodeficiencies, the most severe IDs, all
adaptive immune functions are absent. The condition is fatal unless
the immune system can be reconstituted, either by transplants of immunocompetent
tissue or by enzyme replacement. The immunological, genetic, and enzymatic
characteristics of the diseases show great diversity. SCIDs have
an average frequency of approximately 1 in 75,000 births.
4.1 T-B- SCID
In T-B- SCID both
T and B cells are lacking. To this group of diseases belong RAG1 and
RAG2 deficiencies, Omenn syndrome, reticular dysgenesis, and ADA deficiency.
4.1.1 RAG1 deficiency, RAG2 deficiency and
Omenn syndrome
In recombination activating gene 1 (RAG1) and RAG2 deficiencies and
the Omenn syndrome, recombinase-activating proteins are defective.
These IDs are autosomal
recessive defects. Antibodies as well as T cell receptor and major
histocompatibility complexes (MHCs) have very wide spectrum of specificity,
which originates from combination of coding genes from a large number
of genomic structures. RAGs are crucial proteins in this process by
activating V(D)J recombination in the antibodies and T cell receptor
genes required for generation of the diversity of the recognition sites.
Both RAG proteins are involved in cleaving double stranded DNA during
recombination. T
cells are very low in the patients, but NK cells may appear in
increased numbers.
RAG1 contains homeodomain and a region for RAG2 interaction. RAG2 has
been suggested to be formed of a kelch-like propeller structure and
a PHD domain. RAG2 mutations appear in both regions. In the Omenn syndrome,
recombination is only partially deficient. Some of the Omenn syndrome
patients have mutations that lower the efficiency of RAG interactions.
RAG1 and -2 disruption blocks the initiation of V(D)J recombination
and leads further to complete absence of both mature B and T
cells.
4.1.2 Reticular dysgenesis
Reticular dysgenesis is a very rare autosomal
recessive form of SCID, which generally leads to early death. The
disease is characterized by lack or very reduced levels of B and T
cells, thrombocytes involved in blood clotting, and granulocytes
that form the majority of blood leukocytes, due to the failed maturation
of not only lymphoid but also of myeloid precursor cells.
4.1.3 Adenosine deaminase (ADA) deficiency

Adenosine deaminase (ADA) deficiency accounts for about half of the autosomal
recessive forms of SCIDs.
ADA follows PNP in purine nucleoside catabolism, but deficiency in
this enzyme causes even more severe symptoms than PNP
deficiency, which is a T cell deficiency. ADA degrades toxic adenosine
and deoxyadenosine, which accumulate in the cells of patients. Immature
lymphoid cells are particularly sensitive to these nucleotides. In
addition to immunological defect, most patients with ADA deficiency
also have skeletal abnormalities.
ADA deficiency is an ideal disease for development of gene therapy
methods, because there is selective pressure on nontransduced T cells,
because they are not viable. ADA protein is tolerated also when overexpressed.
The disorder is treated usually with ADA protein substitution therapy
or with bone marrow transplantation.
4.2. T-B+ SCID
In T-B+ SCID T
cells are missing but B cells can be present even in increased
numbers. However, the produced B cells are defective.
4.2.1 X-linked SCID
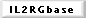
X-linked SCID, accounting for about 50-60% of SCID cases, is caused
by IL-2 receptor γ chain mutations, which lead to very low numbers
of T
cells and NK cells, whereas B cells are present in high numbers.
However, the B cells are immature and defective. IL-2 receptor (IL-2R)
α -chain (CD25) deficiency has also been reported. The γ chain
of the receptor forms part of the receptor also for IL-2, -4, -7, -9,
and -15, affecting the differentiation and growth of lymphocytes. The γ chain
consists of an extracellular domain with WS motif, transmembrane region,
as well as intracellular domain with Box1 and Box2 regions. Some 150
different mutations distributed in all the domains have been determined.
Patients with X-SCID have extreme susceptibility to infections.
4.2.2 JAK3 deficiency

The AR form of SCID is caused by mutations in Janus kinase 3 (JAK3) tyrosine
kinase. IL-2 stimulates the receptor and induces tyrosine phosphorylation
and further activation of JAK3. The γ
c-JAK3 pathway transmits the signal to the nucleus via STATs and effects
the transcription of genes that respond to cytokines. Binding to IL-2
causes dimerization of the receptors and leads into activation of JAK3 tyrosine
kinase. Activated JAK family members phosphorylate multiple tyrosine
residues in the receptors. Signal transducers and activators of transcription
(STATs) are transcription factors, which bind with their SH2 domains
to the phosphotyrosines in the receptor. Activated, dimerized STATs
then dissociate from the receptor and translocate to nucleus, where
they bind to enhancer regions in DNA and thereby effect transcription
of cytokine-responsive genes. Many defective genes can cause similar
symptoms. Mutations in the
γ c chain lead to abrogated or defective JAK3-mediated signal
transduction.
About 30-40% or T-B+ SCIDs are caused by JAK3
mutations. JAK3 is a tyrosine
kinase composed of seven JAK homology (JH) domains. JH1 is the
kinase domain and JH2 a kinase-like module. SCID-causing mutations
have been found from several domains. This seems to be typical for
multidomain signalling proteins since function can be impaired even
when the kinase activity is normal if the enzyme cannot bind effectively
to its substrate(s) or partner(s). Majority of the mutations lead into
truncation or rearrangement of the JAK3 protein from the pseudokinase
domain.
4.3 Other SCIDs
Other combined B and T cell deficiencies include MHC class
I deficiency, which is due to peptide transporter protein 2 (TAP2)
mutations.
4.3.1 TAP2 deficiency
TAP2 transports peptides from the cytoplasm into endoplasmic reticulum,
where MHC I molecules can bind to them. Cells degrade foreign proteins
by proteolysis and generate peptides. Processed peptides bind to MHC
I molecules, which are transported to the cell surface. Then, cytotoxic T
cells recognize the antigen-presenting MHC proteins and kill the
infected cells.
4.3.2 CIITA, RFX, and RFXAP deficiencies
5.1 Wiskott-Aldrich syndrome (WAS)
Wiskott-Aldrich syndrome (WAS) is an immunodeficiency disorder of both
T and B cells characterized by thrombocytopenia, eczema, and recurrent
infections. Patients have progressive lymphopenia. WAS is an X-linked
immunodeficiency. Female carriers are phenotypically normal since the
defective X chromosome is preferentially inactivated. Boys with WAS
have increased serum IgA and IgE levels, whereas IgM is decreased.
T cells are defective and they have fewer microvilli on the cell surface
than normal cells. WAS patients with autoimmune manifestations are
at a high risk of getting malignancies. WAS leads to death within first
two decades of life because of viral or bacterial infections without
bone marrow transplantation.
The gene for WAS has been identified. WAS protein (WASP) interacts
with Cdc42, a GTP-binding protein, that has function in cytoskeleton
reorganization. Defective cell polarization due to WASP mutations could
affect also B
cell T cell interactions. WASP consists of several domains, from
the N-terminus partly overlapping pleckstrin homology (PH) and WASP
homology 1 (WH1) domains, GTPase binding domain, proline rich region
and WH2 domain. The proline rich region interacts with adaptor protein
Nck. More than 100 WAS-causing mutations scattered along the gene and
protein are known. WASP mutations are mainly amino acid substitutions
or nonsense mutations leading to truncation of the protein.
5.2 DiGeorge syndrome
DiGeorge syndrome is a congenital immune disorder characterized by
lack of embryonic development or underdevelopment. Thymic epithelium
is derived from the third and fourth pharyngeal pouches by the sixth
week of gestation. The syndrome is associated with several defects
and it has been called also for CATCH22, because the gene defect is
usually a deletion in chromosome 22, and the symptoms include Cardiac
abnormalities, Abnormal facies, Thymic hypoplasia, Cleft palate, and
Hypocalcinaemia. The degree of thymus problems
varies and only about 20% of the patients have the function or levels
of T cells decreased.
5.3 Ataxia telangiectasia (AT)
Ataxia telangiectasia (AT) is a rare, AR, progressive, neurodegenerative
childhood disease that affects the nervous and other body systems.
The prevalence of AT is in the order of 1:40,000 to 1:100:000 births.
The first signs of the disease usually occur during the first decade
of life. AT means ataxia, lack of muscle control, and telangiectasias
(tiny, red "spider" veins) first in the corners of the eyes
or on ears and cheeks. Immune system is impaired in many patients and
they are also predisposed to leukemia and
lymphoma and they are extremely sensitive to radiation exposure. About
70% of the patients have IgA deficiency and also some IgG subclasses
can be reduced. At the moment there is no cure for AT.
The defective protein, ATM, is a protein kinase that reacts to DNA
damage and affects the accumulation of a tumor suppressor p53, which
is defective in many cancers. ATM mutations delay the accumulation
of p53, allowing cells to replicate without repair of the damaged DNA
and thereby increasing the risk of cancer. Large number of AT-causing
mutations have been identified from different parts of the molecule.
6 Combined immunodeficiencies associated with
other diseases
Combined immunodeficiencies can arise also in association with other
diseases or secondarily to other disorders. A great number of disorders
are related to immunological deficiencies, although the main defect
affects some other functions.
6.1 Down syndrome
Chromosomal defects as Down syndrome, also known as trisomy 21, Turner
syndrome and rings or deletions of chromosome 18 lead into immunodeficiencies.
In Down syndrome, serum IgM is progressively reduced and thymus can
be defective. The patients have facial abnormalities, but also mental
retardation. The triplicated chromosome region contains gene for interferon
receptor and therefore the patients suffering of Down syndrome are
more sensitive to interferons than normal population.
6.2 Abnormalities in chromosome 18
Abnormalities in chromosome 18 may lead to decreased serum IgA
levels and also antibody formation can be impaired.
6.3 Turner syndrome
Turner syndrome patients have generally normal numbers of T and B cells,
but serum IgG
and IgM levels are reduced.
6.4 Chromosomal instability
Chromosomal instability can cause immunodeficiency among other disorders.
6.5 Defective DNA repair
Another cause of immunological symptoms is defective repair of errors
in DNA.
6.6 Fanconi anaemia
| FANCA |
|
| FANCB |
|
| FANCC |
|
| FANCD |
|
| FANCE |
|
| FANCF |
|
| FANCG |
|
| FANCH |
|
Fanconi anaemia is an AR defect of several organs including bone marrow.
T cell functions and serum IgA
levels are decreased.
6.7 Bloom syndrome
Bloom syndrome is marked by chromosomal breakage disorder affecting
multiple systems and characterized primarily by telangiectatic erythema
and short stature. The patients have reduced T cell levels sometimes
accompanied by reduced antibody levels.
6.8 Xeroderma pigmentosum
| XPA |
|
| XPB |
|
| XPC |
|
| XPD |
|
| XPE |
|
| XPF |
|
| XPG |
|
| XPI |
|
Xeroderma pigmentosum and DNA ligase 1 defects are photosensitive dermatoses.
Sunlight causes skin lesions for patients with these diseases.
6.9 ICF syndrome
ICF syndrome has got its name from immunodeficiency, centromeric
instability, facial abnormalities. Serum IgG,
IgM, and IgA levels are usually decreased.
6.10 Nijmegen breakage syndrome
In Nijmegen breakage syndrome microcephaly, short stature and recurrent
infections are typical. Both B and T cell functions are impaired.
6.11 Metabolic defects
Several metabolic defects are accompanied by immunodeficiencies. These
disorders include for example glycogen
storage disease type 1b, mannosidosis, methylmalonic
acidaemia, acrodermatitis
enteropathica, type
1 hereditary orotic aciduria, and transcobalamin
2 deficiency. The degree of immunological problems varies between
and within the diseases.
6.12 Hypercatabolism
Hypercatabolism of immunoglobulins can
also be cause to immunodeficiencies for example in familial hypercatabolism
or intestinal lymphangiectasis.
6.13 Skeletal abnormalities
Skeletal abnormalities in cartilage-hair
hypoplasia and short-limbed
skeletal dysplasia can cause also immunological symptoms.
6.14 Dermatological defects
A number of dermatological
defects affect also T and B
cell responses. These disorders include Netherton
syndrome, dyskeratosis
congenita, and Papillon-Lefèvre
syndrome among others.
6.15 Generalized growth retardation
Generalized
growth retardation is often associated with infections. A number
of these conditions affect the adaptive
immunity.
6.16 Chronic mucocutaneous candidiasis
Chronic mucocutaneous candidiasis patients have persistent cadidial
infections on mucosa and skin. They have severely impaired immunity to Candida.
6.17 Job syndrome
Hyper-IgE syndrome, Job syndrome, is a disorder of neutrophil oxidative
metabolism, typically with extremely high serum IgE
levels, chronic eczema, and recurrent skin and sinopulmonary tract
infections
7. Glossary
Adaptive immunity: acquired
specific immunity.
Autosomal recessive: both
genes at a locus are required to confer a trait in other than X or
Y chromosome.
B cell: antibody producing
blood cell.
Bone marrow transplantation: blood
cells are produced in bone marrow, in the central cavities of bones.
To reconstitute the missing production of blood cells bone marrow can
be transplanted to immunodeficiency patients.
Complement: protein system
that recognizes and kills microorganisms.
Gene therapy: substitution
of a defective gene in patients with a functional one.
Natural immunity: inborn,
non-specific immunity.
T cell: lymphocyte that either
helps or activates other cells (T helper) or kills infected cells (cytotoxic
T cell).
X-linked: inherited by boys
from the mothers in X-chromosome.
|